

Voor rechtstreeks contact belt u
+31 (0)71 528 01 12
Technische documentatie
Unique Device Identifier (UDI) in de EU
Wat is de UDI?
De unieke hulpmiddel-identificatie (Engels: Unique Device Identifier; afgekort UDI) is een unieke numerieke of alfanumerieke code die betrekking heeft op een medisch hulpmiddel. Het zorgt voor een duidelijke en ondubbelzinnige identificatie van specifieke hulpmiddelen op de markt en vergemakkelijkt hun traceerbaarheid, b.v. voor het melden van ernstige incidenten en correctieve acties op het gebied van veiligheid. De UDI bestaat uit de volgende onderdelen:
- een hulpmiddel-identificatie (UDI-DI)
De UDI-DI is een vaste code en is specifiek voor een model of een versie van een hulpmiddel. Dit zijn codes die kunnen worden gekocht bij een aangewezen entiteit.
- een productie-identificatie (UDI-PI)
Dit deel bevat informatie over de productiegegevens, bijvoorbeeld productiedatum, vervaldatum, batch-/lotnummer of een serienummer.
En wat is de Basic UDI-DI?
Binnen de EU is de fabrikant verplicht om aan de hulpmiddelen, samen met een UDI, ook een Basic UDI-DI toe te wijzen. Vanwege hun vergelijkbare namen kan dit een beetje verwarrend zijn. De Basic UDI-DI is gedefinieerd in deel C van bijlage VI van de MDR. De Basic UDI-DI is de primaire identificatie van een hulpmiddel model of een generieke groep hulpmiddelen. Het is de belangrijkste identificatie die wordt gebruikt voor gegevensbestanden in de EUDAMED-database. Er wordt naar de Basic UDI-DI verwezen in technische documentatie, Certificaten voor Vrije Verkoop, EU-conformiteitsverklaringen en samenvattingen van veiligheid en klinische prestaties. De Basic UDI-DI is niet zichtbaar op etiketten/labels of verpakking.
Om een Basic UDI-DI te genereren heeft de fabrikant een contract nodig met een van de door de Europese Commissie aangewezen UDI-uitgevende entiteiten (GS1, HIBCC, ICCBBA of IFA). De meeste entiteiten hebben op hun website tools beschikbaar om de Basic UDI-DI te genereren.
De Basic UDI-DI kan worden gekoppeld aan meerdere UDI-DI’s (bijv. verschillende varianten) die onder dezelfde generieke groep hulpmiddelen vallen, maar een UDI-DI mag uitsluitend worden gekoppeld aan slechts één Basic UDI-DI.
Waar wordt de UDI (UDI-DI + UDI-PI) geplaatst?
De UDI moet op het etiket/label of op het apparaat zelf en op alle hogere verpakkingsniveaus van het apparaat worden geplaatst, met uitzondering van verzendcontainers. De UDI-drager moet leesbaar zijn door ‘Automatic Identification and Data Capture’ (AIDC), maar ook door een persoon (Human Readable Interpretation (HRI)).
Bijvoorbeeld: voor injectiespuiten is een UDI per eenheid vereist, maar wanneer ze worden verkocht als 10 eenheden per doos, is ook een unieke UDI vereist voor deze doos.
Aanvullende eisen
Er zijn aanvullende eisen aan de UDI-DI’s. Als er bijvoorbeeld een maximaal gebruiksaantal voor een product is, moet dit aantal in de EUDAMED-database worden vermeld. Een wijziging van de UDI-DI is vereist wanneer er een wijziging aan het product of anderszins plaatsvindt, die kan leiden tot verkeerde identificatie van het hulpmiddel of onduidelijkheid over de traceerbaarheid ervan.
Datum van toepassing
De verplichting tot UDI-toekenning trad in werking op de datum van toepassing van de twee nieuwe regelgevingen, op 26 mei 2021 voor medische hulpmiddelen en op 26 mei 2022 voor medische hulpmiddelen voor in-vitrodiagnostiek.
De verplichting tot indiening van de UDI-gegevens in de EUDAMED-databank is vastgesteld op 24 maanden nadat EUDAMED volledig functioneel is geworden.
De verplichting tot het plaatsen van een UDI-drager op hulpmiddelen is als volgt vastgelegd:
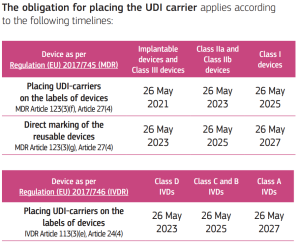
(source: https://health.ec.europa.eu/system/files/2020-09/md_faq_udi_en_0.pdf, accessed on 31-10-2022)
UDI en het kwaliteitsmanagementsysteem
De fabrikant is verplicht UDI’s voor zijn hulpmiddelen toe te wijzen en te onderhouden. Om ervoor te zorgen dat aan de regelgeving wordt voldaan, moeten procedures worden geïmplementeerd in het kwaliteitsmanagementsysteem van de fabrikant. Meer informatie over dit onderwerp vindt u in MDCG 2021-19.